
Why Clean Validation in Pharmaceutical Industry Is Important?
No one can deny the significance of cleaning in the world of pharmaceutical manufacturing, packaging, and distribution. To attain more practical, quality, and potent products, the regulatory bodies have defined, developed, and regulated certain procedures.
The purpose of demonstrating practices is to protect the well-being of consumers and critically lower the chances of cross-contamination. That’s the reason such practices have substantially evolved in this sector over the last two decades. This article ‘Why clean validation in the pharmaceutical industry is important’ is a comprehensive guide to essential key points of validation, including general description, key types, and common challenges. Let's start.
1.What is Cleaning Validation in Pharmaceuticals?

Cleaning Validation in Pharmaceuticals- Picture courtesy: GxP Cellator
Cleaning validation in pharmaceuticals is an effective practice implemented to reassure cleaning methods. This is followed by eliminating the residues or leftovers in pharmaceutical equipment from previously used ingredients. Such as active pharmaceutical ingredients, excipients, etc., and the production process has encountered strict regulatory approaches. This involves systemically work patterns with clean and hygienic handling to maintain consistent manufacturing batches free from cross-contamination up to approved and acceptable limits.
Performing these fundamental practices can safeguard consumers against harm. That’s the reason it is mandated by international regulatory bodies such as the Food and Drug Administration (FDA), World Health Organization (WHO), and European Medicines Agency (EMA). Therefore, essential performing highlights are validated by Good Manufacturing Practices (GMP). Moreover, it is critical to follow up if compromised; it may result in trace residues in the drug, which may exaggerate harmful and toxic effects imposed on other medicines.
2.Why Cleaning Validation is Crucial in Pharmaceutical Manufacturing?
Significance of clean validation in pharmaceutical industry- Picture courtesy: McMaster
To driving cleaning validation in pharmaceutical manufacturing can support the following points:
Cross Contamination Prevention
The clean validation protocols document that equipment can be processed safely. There won't be doubts about fundamental traceability. So, equipment won't be the source of cross-contamination, which is significant to evaluate when it comes to formulating different products in the same machine.
Product Quality & Patient Safety
Pharmaceutical formulations are pure if they don’t contain unintended substances. Therefore, it directly improves the quality. Adhering to your equipment and procedure with cGMP will proportionately increase effectiveness throughout the working environment. Patient safety is the foremost priority of the manufacturer. Thus, cleaning validation protocols provide safety to consumers from mishaps.
Regulatory Compliance
The thorough execution of regulatory compliance in the protocols of the pharmaceutical industry helps in sustaining quality. Therefore, the major challenges such as batch withdrawal, losing customers' trust, and uncertainties connected with penalties will no more.
Risk Reduction & Cost Efficiency
Globally, the pharmaceutical industry aims to offer products at a budget. Following clean validation protocols always helps minimizing unwanted risks of basic failures, batch withdrawals, and regulatory challenges. Thus, can streamline the usual working with a good reputation of the brand for long-term profit.
3.Regulatory Guidelines for Cleaning Validation
Regulatory bodies, for example, the FDA, EMA, ICH, and WHO, have each created a set of specific regulatory guidelines for effective cleaning validation. Listed below are a few of the most important global standards.
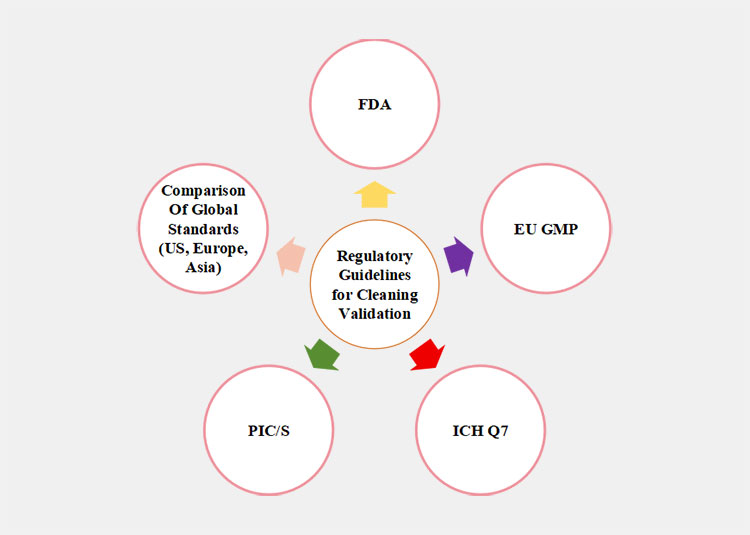
Regulatory Guidelines for Cleaning Validation
FDA

Food and Drug Administration (FDA)
The Food and Drug Administration (FDA) in the United States controls the manufacturing and distribution of therapies; thus, it has devised the comprehensive regulatory guidelines for cleaning validation. Like in FDA 21 CFR Part 211.67, it outlines key practices of cleaning responsibility to suitable personnel, and maintenance of a cleaning schedule.
For inspection and evaluation of cleaning processes, manufacturers are obligated to devise a definition of an acceptable level of cleanliness. Furthermore, according to FDA guidelines, pharmaceutical developers are obligated to maintain a record of sampling methods, like swabbing or rinsing, as well as direct surface evaluations.
EU GMP

European Medicines Agency -Picture Courtesy: Scendea
Pharmaceutical products manufactured in the European Union are assessed and tested for safety by the European Medicines Agency (EMA) Good Manufacturing Practice. This agency has issued guidelines for designing, implementing, and validating cleaning protocols. These directives require manufacturers to establish Health-Based Exposure Limits (HBELs) for every drug formulation and reevaluate these limits during the product lifecycle.
In addition, EMA EudraLex Volume 4, Annex 15 also describes expectations for conducting risk assessments for cleaning validation, which means not just visually clean matters, but keeping proper documentation, records, and defining scientifically-determined acceptance criteria must be applied.
ICH Q7

ICH Q7
ICH Q7A recommendations for cleaning validation are identical to those in the FDA GMP guidelines. Both ICH Q7A and the FDA draft guidance require the matrix approach for product selection in cleaning validation. By grouping equipment of similar design, structure, and working principle, this method reduces the need to validate each instrument separately, while ensuring that cleaning steps are effective in preventing cross-contamination between batches.
As per ICH Q7A guidelines, manufacturers must apply swabbing, rinsing, or alternative methods for sampling. Also, developers are advised to develop practical and achievable residue limits depending upon the toxicity of the residue and the physiological/pharmaceutical activity of the API.
PIC/S

PIC/S- Picture Courtesy: PharmOut
Effective from July 1, 2018, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) adopted cleaning validation principles that are fully aligned with those established by EMA. This harmonization leads to consistent global regulatory compliance across 50 member countries. So, PIC/ member countries are mandated to define HBELs in shared facilities and follow risk-based cleaning validation. Drug regulatory agencies in these countries evaluate the compliance of manufacturing facilities as per EU standards.
In addition, these guidelines refer to AIDE-MEMOIRE, a comprehensive resource detailing cleaning validation expectation. This document is an essential read for manufacturers who are striving to maintain compliance and to avoid regulatory challenges.
Comparison Of Global Standards (US, Europe, Asia)

Comparison Of Global Standards of Cleaning Validation- Picture Courtesy: Uventia
Here is a comparison of global standards of cleaning validation across continents, like the US, Europe, and Asia.
| Features | United States | Europe | Asia |
| Primary Authority | FDA | EMA or EU GMP | In Asia, regulators generally align with ICH Q7 and PIC/S guidelines. |
| Regulatory Stance | Science and risk-based and use solid validation evidence | Adopt a risk-based assessment that is reassessed throughout the medicine lifecycle | Emphasize the same risk-based validation studies as recommended by EMA. |
| Acceptance Limits | Have to be scientifically justified as per the severity of residue and the pharmacology of AP. | EMA guidelines explicitly state to specify HBELs. | Comply with ICH/PIC/S, thus have to define HBEL. |
| Sampling Methods | Swab and rinse or alternatives | Swab & rinse | Same as the USA, includes swab, rinse, or alternative approaches |
| Visual Cleanliness | Necessary but not adequate alone | Required; however, it is not the sole cleaning criterion | Must be implemented, but cleaning must be assessed by other methods as well. |
| Documentation | Thorough SOPs, cleaning protocols, and written procedures mentioning date, time, materials, and responsibilities. | Comprehensive record-keeping of every cleaning validation activity | Prepare documentation as per the guidelines of ICH/PIC/S, but formatting may vary |
4.Key Elements of an Effective Cleaning Validation Program
Evaluating the effectiveness of a cleaning validation program can be complex and demanding, with numerous points of potential failure. However, by understanding key elements of an effective cleaning validation program, manufacturers can minimize errors and satisfy worldwide regulatory expectations:
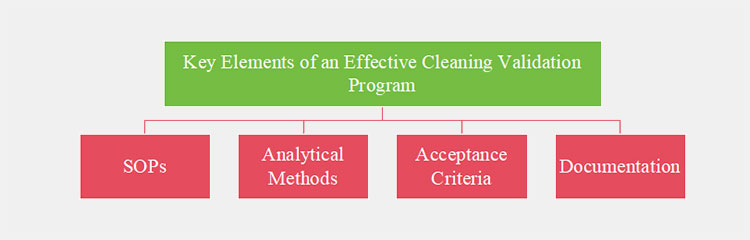
Major Points of Clean Validation in Pharmaceutical Industry
SOPs

SOPs- Picture Courtesy: WCA
Clear, detailed, and well-written Standard Operating Procedures (SOPs) form the backbone of a cleaning validation program. They are step-by-step instructions or written procedures that guide personnel responsible for cleaning equipment both in industrial and laboratory settings.
Generally, the SOPs outline scope and objectives for cleaning validation, responsibilities of personnel, definition of technical terms, prerequisites for validation activities, meticulous cleaning procedure, methodologies, operator competency standards, and steps to revalidate cleaning methods.
Analytical Methods

Analytical Methods- Picture Courtesy: Sciex
To prove the effectiveness of cleaning validation, manufacturers use multiple analytical methods and tools like High-Performance Liquid Chromatography (HPLC), Gas Chromatography (GC), Total Organic Carbon (TOC), or UV spectroscopy. With the aid of these tools, manufacturers can quantify the amount of drug product residue or cleaning substance left on equipment after cleaning.
This quantification provides an independent assessment that the equipment they’re using to produce the product is clean and is ready to process the next batch. However, before implementing any analytical method, manufacturers are required to validate their preferred analytical method and establish its sensitivity towards the target analyte.
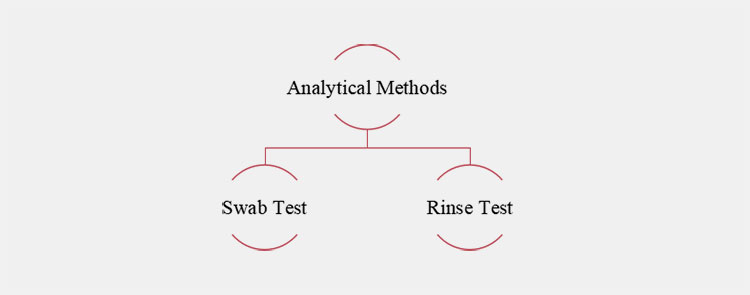
Major Testing of Analytical Methods
Swab Test
A relatively direct sampling method in which surfaces of equipment are wiped with uses clean and pretreated swab. The collected samples are examined to determine residue levels. This test is useful for dried residues or analytes that are insoluble in rinse solvents. As it provides localized results, it is effective for sampling accessible and hard-to-access surfaces.
Rinse Test
On the other hand, the rinse test is an indirect sampling method in which water or other solvents are used to wash clean equipment. Then the said solution is analyzed for contaminants. Using this test, larger surface areas, inaccessible devices, or components that can’t be normally disassembled are sampled and appraised.
Acceptance Criteria

Acceptance criteria- Picture Courtesy: Smart Maker
One of the most critical elements to determine the effectiveness of the cleaning validation program is the acceptance criteria. If this minimum accepted residue limit is defined accurately, manufacturers can validate their cleaning validation with greater confidence. Typically, acceptance criteria are categorized into three diverse testing parameters:
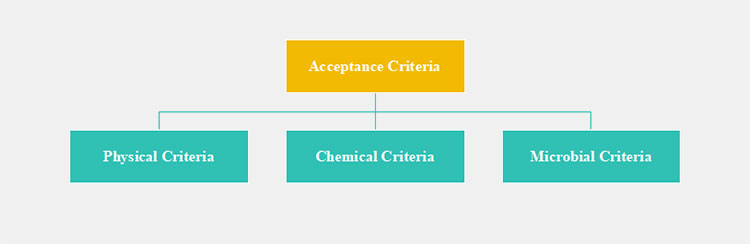
Acceptance Criteria Parameters
Physical Criterion: Typical acceptance limit for physical assessment is that there shouldn’t be any noticeable stain, spot, residue, or films of the previous product or cleaning agent.
Chemical Criterion: Residue levels of active pharmaceutical ingredients, excipients, and other chemicals must be detected below scientifically justified limits. Generally, one product should not be spotted more than 10 ppm (parts per million) in another formulation.
Microbial Criterion: It is vital that equipment is devoid of any microbial contamination. For example, bacterial count after sterilization or cleaning should not be greater than 20 CFU (colony-forming units).
Documentation

Documentation- Picture Courtesy: BioPharma Service
In manufacturing, each process and procedure of cleaning validation must be supported by comprehensive documentation and precise record-keeping. For regulatory compliance, there must be a clear and traceable trail from the design of cleaning protocols, through their execution, and to the ultimate acceptance of sampling and analytical methods.
Manufacturers, developers, and engineers should ensure detailed documentation of written protocols of cleaning validation, raw analytical data inputs, chromatograms from HPLC and GC testing, sampling logs, records of cleaning agents’ utility, errors detected, and CAPA findings, revalidation triggers, cleaning schedule, and residue levels detected after cleaning.
5.Common Challenges in Cleaning Validation
Cleaning validation in pharmaceutical industry faces several challenges as it is a very sensitive and crucial process. Some of the challenges are outlined below.
Complex Equipment

Complex Equipment- Picture Courtesy: ALFA laval
Cleaning validation particularly becomes challenging when the equipment exhibits complex design, recessed areas, and multiple interconnected parts. These structural intricacies establish opportunities for residues to persist, consequently undermining sterility and product safety.
For optimal mitigation of this challenge, it is crucial to implement targeted cleaning protocols along with specialized equipment and precise inspection methods. Continuous in-process monitoring and documented risk assessments ensure successful execution of cleaning validation and GMP protocol compliance.
High-Risk APIs (High-Potency Active Ingredients)

High-Risk APIs (High-Potency Active Ingredients)- Picture Courtesy: Qingmu Pharmaceutical
High-risk active pharmaceutical ingredients (APIs), particularly high-potency active pharmaceutical ingredients (HPAPIs) create major challenges in cleaning validation due to their strong pharmacological effects and low residue limits, even minute residues increase contamination risk and undermine product quality.
To overcome these challenges, cleaning procedures must be specifically designed considering the toxicological properties of each HPAPI. This includes the incorporation of specialized equipment, as well as highly specific and precise analytical methods for residue detection. For efficient cleaning validation of high-risk APIs, it is crucial to perform in-process monitoring along with accurate documentation and risk assessment.
Test Sensitivity and Validation Costs

Test Sensitivity and Validation Costs- Picture Courtesy: Medical Device Network
Highly sensitive analytical methods are a prerequisite for cleaning validation, as they ensure detection of even trace residues that could result in degradation of product quality and safety. The sensitivity level required for cleaning validation depends upon the toxicological properties and clinical dose of the product.
In cases of high-potency APIs, extremely low residue limits are required because even a trace amount can harm the subsequent product.
To address these challenges, it is important to implement risk-based approaches. Cost-effective analytical alternatives such as TOC and UV can be applied where necessary, while maintaining compliance with GMP and regulatory protocols.
6.Best Practices for Successful Cleaning Validation

Best Practices for Successful Cleaning Validation- Picture Courtesy: Zamann Pharma
Implementation of effective practices is crucial for successful cleaning validation process of pharmaceutical equipment. Some of the best practices are outlined below.
Risk-Based Approach

Risk-Based Approach- Picture Courtesy: DCAT
A risk-based approach emphasizes evaluating and controlling the potential product risk during the manufacturing process. The process begins with calculating the Maximum allowable carryover (MACO) using toxicological data, therapeutic dosage information and product-specific properties to define the acceptable residue limit. Further parameters like equipment design, cleaning challenges and microbial residues are evaluated for optimal efficiency and patient safety.
Periodic Revalidation

Periodic Revalidation
Periodic revalidation ensures that the cleaning procedures are compelling regardless of routine use and process modifications. The revalidation interval is determined after risk assessment, considering other parameters such as product potency, cleaning challenges, toxicity and manufacturing intricacy. Additional circumstances such as introduction of new products, equipment design alterations or revised regulatory framework mandate the execution of revalidation action. A productive revalidation procedure starts with a framework, undertaking risk assessment, execution, data analysis and accurate documentation.
CIP/SIP
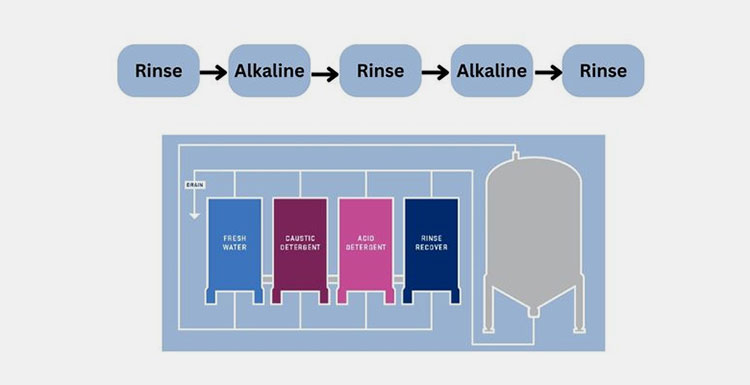
CIP/SIP- Picture Courtesy: Future Generation
A clean-in-place (CIP) and steam-in-place (SIP) are significant procedures, involving cleaning and sanitizing the pharmaceutical equipment without the need to disassembling them.
CIP deploys automated cycles to remove product residues and microbial counts from internal surfaces of equipment using water, detergents and cleaning agents. While SIP is an extension of CIP in which hot steam is used to clean the pharmaceutical equipment. This process reduces the risk of contamination while enhancing sterility level and product safety. Other key parameters such as temperature, cleaning agent concentration and flow rate are continuously monitored for effective cleaning.
Staff Training and Audits

Staff training and audits- Picture Courtesy: Rasayanika
Functional and reliable cleaning validation relies on competent and well-trained staff. Influential training should be reinforced along with proper demonstration and continuous assessments. Internal audits are implemented to verify compliance with procedures, identify potential gaps and monitor the efficacy of cleaning validation process. Meanwhile, external audits provide an additional evaluation oversight. Audit findings and remarks must be accurately documented and addressed through corrective and preventive actions (CAPA) to attain continuous improvement.
7.Future Trends in Cleaning Validation
Future trends in cleaning validation are reshaping pharmaceutical methods of maintaining safety standards. Some of them are as follows.
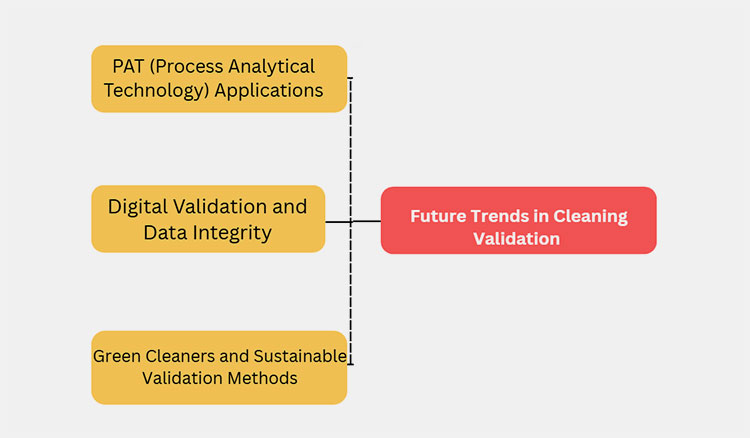
A Flow Diagram Illustrating Future Trends in Cleaning Validation
PAT (Process Analytical Technology) Applications
PAT- Picture Courtesy: BRUKER
Future trends in cleaning validation are evolving towards automated methods to attain a real-time monitoring approach. PAT transitioned conventional or old validation techniques to inline and online tools such as spectroscopic and chromatographic analyzers, near-infrared (NIR). This is based on sensors that detect residues left on equipment surfaces immediately. This paradigm shift by PAT in cleaning validation reduces errors and downtime and also complies with pharmaceutical regulatory requirements.
Digital Validation and Data Integrity

Digital Validation and Data Integrity- Picture Courtesy: Kneat
Digital validation and data integrity are strategically important future trends that ensure compliance with data integrity protocols because traditional paper-based documentation is labor-intensive. Additionally, it is more inclined towards errors and prone to challenges during inspections and monitoring. These technologies applications in cleaning validation consist of electronic batch records (EBR), audit trails, IoT sensors and PAT tools, and cloud-based validation management.
Green Cleaners and Sustainable Validation Methods

Green Cleaners and Sustainable Validation Methods- Picture Courtesy: ColoroxPro
Green cleaners and sustainable validation methods in cleaning validation are rising factors, as they emphasize the use of eco-friendly biodegradable cleaning agents and aqueous-based cleaning solutions. With an increase in environmental concerns, the pharmaceutical industry is increasingly adopting green chemistry and sustainable cleaning approaches. This trend underlines scalable validation methods such as eco-friendly, non-toxic cleaning detergents and solvents, minimal water and energy usage, and reduced waste disposal.
Conclusion
Clean validation in the pharmaceutical industry is a foremost element that may be underestimated, though a critical one if not taken seriously for medical equipment, pharmaceutical machinery, and the workplace. Inadequate practices in handling pharmaceuticals may not only result in non-conformities but also put the lives of patients at higher risk. Therefore, companies must be compliant with all comprehensive clean validation strategies to produce quality products and bring safe medication to patients. By this, the future of the pharmaceutical industry can be boosted with high trust, effectiveness, and profitability. Reach out to us to revolutionize the cleaning validation of your company!
Don't forget to share this post!
Capsule Filling Machine Related Posts































Capsule Filling Machine Related Products




-Empty-Capsule")



Capsule Filling Machine Related Videos
CONTACT US
Tell us your raw material and project budget to get quotations within 24 hours.
WhatsApp Us: +86 156 0710 8630

Want the best price & newest pharmaceutical machinery buying guide,tips and trends sent straightly to your box?Sign up for AIPAK’s monthly newsletter,we’re free for your consultation and Offer you the most suitable solutions!
The Buyer's Guide
- Capsule Filling Buyer's Guide
- Blister Packaging Buyer's Guide
- Tablet Counting Buyer's Guide
- Tube Filling Buyer's Guide
- Cartoning Buyer's Guide
- Gummy Making Buyer's Guide
- CO2 Extraction Buyer's Guide
- Empty Capsules Buyer's Guide
- Suppository Filling Buyer's Guide
- Tablet Coating Buyer's Guide
- Tablet Press Buyer's Guide
- Softgel Encapsulation Buyer's Guide
Most Popular
- 7 Importance Of Pharmaceutical Packaging In Different Applications You Must Know
- 6 Advantages You Must Know About Tablet Counting Machine
- 8 Advantages of Blister Packaging You Must Know
- 6 Critical Applications of Automatic Capsule Filling Machine
- 6 Stations You must Know to Improve the Filling Quality of Automatic Capsule Filling Machine
 Tell us your material or budget,we'll reply you ASAP within 24 hours
Tell us your material or budget,we'll reply you ASAP within 24 hours